Multiple scattering (MS) theory is widely used to calculate physical
properties of solids, ranging from electronic structure to optical and
X-ray response. X-ray absorption spectroscopy (XAS) uses
energy-dependent modulations of photoelectron scattering to probe
excited states and thus is important for determining electronic and
chemical information from X-ray spectra. XAS is usually divided into
the extended X-ray absorption fine structure (EXAFS) with photoelectron
energies above ~ 70 eV, and the X-ray absorption near edge structure
(XANES) in the 0-70 eV range.
Theoretical calculations of photoelectron scattering are now
an integral part of both EXAFS and XANES analysis. These theoretical
calculations have grown in sophistication and complexity over the past
twenty years. Fortunately computing power has increased dramatically
(in accordance with Moore's law) during the same time period, and as a
result EXAFS calculations are now fast, accurate and easily executed on
inexpensive desktop computers.
The X-Ray Absorption code has been parallelized.
 |
Why Parallelize Computational X-Ray Absorption?
In contrast to EXAFS, XANES calculations are even today time consuming
for many materials. The photoelectron mean free path is large at the low
photoelectron energies of the XANES region, so accurate XANES calculations
require large atomic clusters and remain challenging on even the fastest single
processor machines. Furthermore, the photoelectron scattering is strong for
low energies, so that full multiple scattering calculations are required.
These calculations require repeated inversions of large matrices which scale
as the cube of the size of the atomic cluster. Fortunately, parallel
processing using the message passing interface (MPI) standard, combined with
modern Lanczos type MS algorithms, can speed real-space XANES and electonic
structure calculations by about two orders of magnitude. In particular,
Feff, one of the most commonly used programs for XAS analysis (developed
at the University of Washington) has been improved in this manner, leading
to a parallel version, FeffMPI.
|
|
How is the Parallelization Realized?
A series of similar MS calculations must be done at a large number
(typically of order 100) of energy points to obtain a complete XANES
spectrum. This number is determined by the natural energy resolution
(due to lifetime broadening and inelastic losses) and the range of the
XANES region (typically below 30 eV of threshold) for which full MS
calculations are needed. Thus it is reasonable to consider doing these
similar MS calculations in parallel. Since we aim to model the physical
process of x-ray absorption, it is natural to exploit the intrinsic
task parallelism (or physical parallelism) in this problem, namely that
the x-ray absorption at a given x-ray energy is independent of the
absorption at other energies, assuming they are separated by the inherent
energy resolution (typically a fraction of an eV). Thus a natural way
to parallelize the full spectral calculation is simply to distribute the
energy points among an available set of processors. We can then assemble
the results to obtain the full absorption spectrum.
To this end we have developed a parallel version of the ab initio full
MS XANES code Feff8 using the Message Passing Interface (MPI) standard.
This parallel code (here dubbed FeffMPI) compiles and runs without changes
on all currently available operating systems tried to date (e.g., Linux,
Windows NT, Apple Os X, IBM-AIX, SGI, CRAY ....).
The starting point for parallelizing any code is to determine which
parts of the calculation are the most time consuming. Profiling tests
showed that only a small section (about 100 lines that call matrix
inversion routines) of the code accounted for about 97% of the total
runtime in typical calculations. Altering these calculations to run
in parallel is straightforward with MPI, because each step involves similar
calculations and utilizes identical matrix inversion routines.
|
|
What is the Performance of the Parallel Code?
FeffMPI runs on PCs running Windows and Linux as well as most commercial
UNIX vendor machines. The list of machines supported has recently been
extended to include the Apple Macintosh running the new OS X operating system.
FeffMPI is now also operating on parallel processing clusters at the University
of Washington and at DoE's National Energy Research Scientific Computing Center
(NERSC). In fact, FeffMPI compiles and runs without changes on all currently
available operating systems tried to date (e.g., Linux, Windows NT, Apple Os X,
IBM-AIX, SGI, CRAY ....) with speedups of up to 30. A speedup of 30
makes it possible for researchers to do calculations they only dreamed
about before. One NERSC researcher has reported doing a calculation in
18 minutes using FeffMPI on the NERSC IBM SP2 cluster that would have taken
10 hours before. In 10 hours this researcher can now do a run that
would have taken months before, and hence would not have been even
attempted.
|
|
|
|
With the improved efficiency of FeffMPI now in hand, it is
feasible to carry out XANES calculations which otherwise would
have been impractical. For example, a few days of calculations
on a 48 processor Linux cluster can now complete a calculation
that would take a year on a current single processor. Systems
such as complex minerals, oxide compounds, biological structures
and other nano-scale systems are obvious targets for this type
of improved capability. The improved speed should be very
useful, for example, for magnetic materials, which often have
a large number of inequivalent sites of absorbing atoms,
requiring many separate calculations to produce a full XANES
or XMCD (X-ray magnetic circular dichroism) spectrum. Finally,
the availibility of rapid calculations now permits closed loop
fitting of XANES spectra both to physical and chemical
phenomena.
As one example of these calculations, we show how XANES calculations can
be used in the study of amorphous germanium (aGe). It is well known
that the structure of amorphous tetrahedral semiconductors can be
modeled well by an approach called a continuous random network (CRN).
In this method, the amorphous semiconductor retains the parent structure
of the crystal, but various degrees of freedom, the interatomic distance,
the bond angle and the dihedral angle, are allowed to become statistically
disordered. Originally built by hand with ball-and-stick models, CRNs
have been generated by computer, and the degree of disorder in the structural
parameters is determined by energy minimization methods. Comparisons of CRN
models with EXAFS data have been done, but these comparisons
were not extended into the XANES region because of the inability to perform
ab initio XANES calculations, and even in the EXAFS region the
calculations were limited to a simple single scattering theory.
Here we show that we can use FeffMPI and a CRN model to reproduce the main
features in the XANES of crystalline and amorphous germanium.
As a starting point for the XANES calculation of aGe, we first
modeled the XANES of crystalline germanium to determine the cluster
size needed to accurately reproduce the XANES. We found that a
cluster of 87 atoms, which includes the first 7 coordination shells,
out to a distance of approximately 0.78 nm is sufficient to
reproduce the main features of the experimental data.
The aGe XANES calcuations were then carried out using similar clusters
of 87 atoms that had nearly the same size as the crystalline cluster,
because the CRN yields a structure that has the same density as
crystalline Ge, to within a few percent. In order to get a good
ensemble average over the inequivalent sites of the CRN, we ran
the same Feff calculation over a total of 20 sites in the CRN. We
tested the calculation on a single processor desktop machine, where a
single run took approximately one hour. We then used a 16 processor
cluster where each calculation took about 3 minutes. Using FeffMPI
and this fairly modest cluster size thus reduced the total
calculation time from what would have been 20 hours on the
desktop machine to 1 hour, for a 20-fold improvement over the
single processor desktop system and in agreement with our
previous result that moderate-sized clusters (approximately 33
processors) typically give a 20-fold spped increase compared
with an equivalent single-processor system, and up to a 50-fold
increase compared with typical single-processor desktop
systems.
In Figure 1(a) we show the 87 atom cluster used to calculate the XANES
of crystalline Ge. In Figure 1(b) we show a similar cluster of 87
atoms of aGe from the CRN displayed with the same length
scale. As shown in the figure, each cluster is about 1.5 nm across.
In Figure 2 we show the full 519 atom cluster of aGe from the CRN with
a typical cluster of 87 atoms highlighted in the interior. Although there
are several hundred atoms in the interior of the 519 atom cluster that are
fully coordinated by 87 atoms, we obtain an accurate ensemble average
using just 20-30 atoms near the center of the cluster. The convergence occurs
quickly since averaging over N sites includes 4N first neighbor
atoms, 12N second neighbor atoms, etc. The disorder in the CRN is large enough
that the separation of the neighboring atoms into separate coordination shells
breaks down by the third or fourth shell.
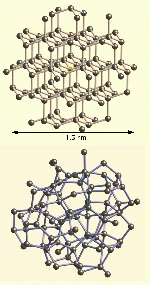 |
| Figure 1:
(a) The 87 atom cluster used to calculate the XANES of crystalline Ge. (b) A similar cluster of 87 atoms of aGe from the CRN displayed with the same length scale.
|
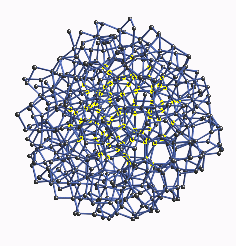 |
| Figure 2:
The full 519 atom cluster of aGe from the continuous random
network with a typical cluster of 87 atoms highlighted in the interior.
|
|
Papers/Presentations
|
Alex L. Ankudinov, Charles E. Bouldin, John J. Rehr, James S. Sims and Howard K. Hung, Parallel Calculation of Electron Multiple Scattering Using Lanczos Algorithms,
Physical Review B, 65
(10)
,
2002.
Links:
postscript and pdf.
|
|
Charles E. Bouldin, James S. Sims, Howard K. Hung, John J. Rehr and Alex L. Ankudinov, Rapid Calculation of X-ray Absorption Near Edge Structure Using Parallel Computing,
Journal of X-ray Spectroscopy, 30,
2001,
pp. 431-434.
|
|
James S. Sims, William L. George, Steven G. Satterfield, Howard K. Hung, John G. Hagedorn, Peter M. Ketcham, Terence J. Griffin, Stanley A. Hagstrom, Julien C. Franiatte, Garnett W. Bryant, W. Jaskolski, Nicos S. Martys, Charles E. Bouldin, Vernon Simmons, Olivier P. Nicolas, James A. Warren, Barbara A. am Ende, John E. Koontz, B. James Filla, Vital G. Pourprix, Stefanie R. Copley, Robert B. Bohn, Adele P. Peskin, Yolanda M. Parker and Judith E. Devaney, Accelerating Scientific Discovery Through Computation
and Visualization II,
NIST Journal of Research, 107
(3)
,
May-June, 2002,
pp. 223-245.
Links:
postscript and pdf.
|
|
James S. Sims, John G. Hagedorn, Peter M. Ketcham, Steven G. Satterfield, Terence J. Griffin, William L. George, Howland A. Fowler, Barbara A. am Ende, Howard K. Hung, Robert B. Bohn, John E. Koontz, Nicos S. Martys, Charles E. Bouldin, James A. Warren, David L. Feder, Charles W. Clark, B. James Filla and Judith E. Devaney, Accelerating Scientific Discovery Through Computation
and Visualization,
NIST Journal of Research, 105
(6)
,
November-December, 2000,
pp. 875-894.
Links:
postscript and pdf.
|
|
|
|