Parallel Cooperative Algorithms for Protein Folding
John Moult ([email protected])
Jan T. Pedersen ([email protected])
The elucidation of protein structure plays a quintessential
role in the development of our understanding of
evolution and the function of biological processes.
Currently protein structures are determined experimentally
by x-ray crystallography and NMR spectroscopy. These methods,
although accurate, are time consuming and suffer from
inherent drawbacks. There is a need for the development of
theoretical methods which allow for the ab-initio
calculation of protein structure.
It is important that the elucidation of protein structure
can be performed at the same rate as sequencing of genomes.
The phenotype (product) of a gene is a protein, and the pressure
of natural selection is on the phenotype and not on the DNA.
In order to be able to understand the meaning of large
amounts of genomic data we need to understand how proteins
(enzymes etc.) create specific functional structures.
The Protein Folding Problem refers to the combinatorial
problems involved in enumerating the conformations of a given
Protein molecule. Cyrus Levinthal (1968) outlined
this in a simple paradox: Let each amino-acid residue
in a 100 residue protein have 6 possible conformations, this
leads to 6^100 possible conformations available for this
protein, this calculation does not include sidechain conformations
which will increase the number of degrees of freedom further.
The question is now how does the protein fold given this large
number of possible conformations. These simple calculations
urge the development of new efficient and accurate search methods.
This project attempts to address the two main problems involved
in the protein folding problem. The first problem is
the understanding of the energetics involved in protein
folding. This is here addressed from a thermodynamic view
point, developing empirical Force Fields parameterised
using experimentally determined protein structures. The second
problem deals with the search problem outlined in
Levinthals Paradox above.
The developed search methods and force-fields are applied
to simulations of fragments of proteins which are known
to contain structure, independently of the rest of the
structure (Early Folding Units (Wetlaufer 1973)),
and to small peptide chains that have been shown by nuclear
molecular resonance (NMR) have a structure in solution.
References
Cyrus Levinthal
Are there pathways in protein folding ?
J. Chim. Phys. (1968), vol 65, pp 44-45
D.B. Wetlaufer
Nucleation, Rapid Folding, and Globular Interchain Regions
in Proteins
Proc. Natl. Acad. Sci. (1973), vol 70, pp 697-701
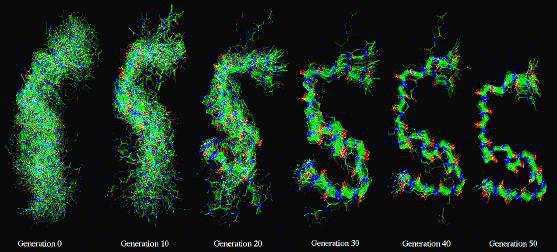
Figure 1
An illustration of Genetic Algorithm (GA) simulation of a small 22
Amino Acid peptide corresponding to the membrane binding domain
of Blood Coagulation Factor VIII. The simulation attempts to
select the most likely conformation of the peptide using an
objective energy function which describes the mechanics of the
peptide chain. The Genetic Algorithm emulates the process of
natural selection in order to search the energy space
efficiently. This is illustrated in the figure by a set of
snapshots taken during a 50 generation simulation, it is seen
how, gradually, one conformation is selected above the others
through cross-overs and recombination of fragments within the
population of structures. The GA search method is able to exploit
parallel computing resources to the full. The above simulation is
performed using the 19 node IBM SP2 at NIST.
A PostScript version is available.

Figure 2
Structure of Blood Coagulation Factor VIII membrane binding
domain. The structure is the best obtained in a series of Genetic
Algorithm simulations. The structure shown is similar to that
obtained through nuclear molecular resonance (NMR) experiments.
The peptide is shown in two representations; A) CPK illustrating
the compact nature of the molecule, B) Stick rendering, showing
the chemical composition of the peptide.