
Walter J. Stevens ([email protected])
Recombinant DNA technology and genetic engineering have progressed to the point where changing the atomic or amino acid composition of a protein is a routine activity in any modern molecular biology laboratory. For a decade or more, scientists have dreamed of being able to use molecular biological methods to create or alter proteins in a deliberate, predetermined way in order to give them desirable and useful properties. The use of such "protein engineering" methods to modify and optimize biological molecules (biomolecules) for use in technological applications such as chemical manufacturing, bioremediation, and synthetic materials requires a detailed understanding of how biomolecular structure relates to function. Today, structures of proteins are being determined at a very rapid rate by X-ray crystallography and NMR spectroscopy, but the models and computational methods that predict structure from amino acid (atomic) composition or relate structure to function are still in the early stages of development.
This project is aimed at developing computational methods that predict how the structure of an enzyme (a protein that catalyzes a chemical reaction) is related to its catalytic activity. The project is being carried out at the Center for Advanced Research in Biotechnology (CARB), which is a joint research center of NIST and the University of Maryland Biotechnology Institute. The research at CARB is focussed on the determination of protein structures and developing and understanding structure/function relationships.
Enzymes have many potential uses in chemical technology, but naturally occurring enzymes may have to be modified in order to effect the desired reactivity. Ideally, the desired modifications would be "designed" by using predictive models that relate the structure to the reaction. Predicting the behavior of any chemical reaction requires the use of quantum mechanical models that are able to describe the making and breaking of chemical bonds. Great strides have been made over the past twenty years in computational "quantum chemistry", but the average enzyme is so large (thousands of atoms), that the direct application of quantum chemistry methods would be prohibitively expensive even on the largest computers. However, examination of the structures of typical enzymes and their complexes with other molecules reveals that typically only a small portion of the enzyme is actually in contact with the reactants or potentially involved in the reaction. The bulk of the enzyme supports the exquisitely-designed catalytic "active site" or may be involved with other biological functions such as the control of the catalytic activity in living systems.
The computational approaches being developed in this project are aimed at applying quantum chemistry to an enzyme active site and the chemical reactants, while treating the bulk of the enzyme and the solvent as "perturbing" parts of the system that don't have to be described quantum mechanically because they are not directly involved in the enzymatic chemistry. Even with this simplifying assumption, the average enzymatic active site is large enough to constitute a major challenge for existing quantum chemistry computer programs. Thus, the quantum calculations are being done with programs that have been modified and optimized for use on parallel computers. The particular program that is being used to develop the new methods for handling bulk protein and solvent is GAMESS (Generalized Atomic and Molecular Structure System) which is written and maintained by the research group of Professor Mark Gordon at Iowa State University ([email protected]).
The new methods are based on the use of "effective fragment potentials" (EFP) in the quantum mechanical Hamiltonian that provide the coupling between the chemically active part of the system and the remaining protein and solvent. The EFP contain terms which model coulombic interactions, polarization, and local approximations of exchange repulsion at the interface between the quantum region and the bulk.
Figures 1 and 2 are graphical representations of an enzymatic system that is currently under study as a test case for the new methods. The enzyme is bovine pancreatic ribonuclease A (RNase) which is one of the most widely studied enzymes for the past forty years. RNase is a digestive enzyme that catalyzes the hydrolysis of single-stranded RNA. Many high resolution X-ray structures are available for RNase and RNase/substrate complexes, which makes it an ideal candidate for verifying modeling methods.
Figure 1
A PostScript version is also available.
Figure 2
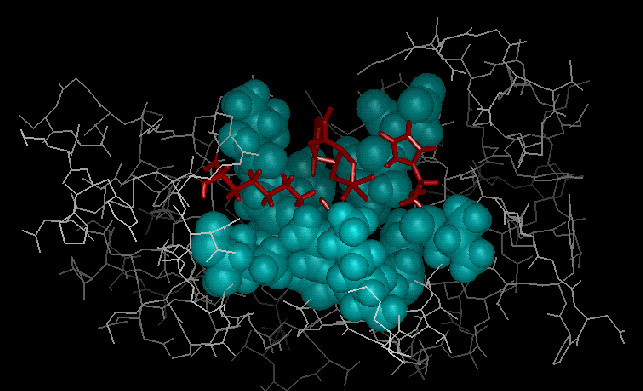
A PostScript version is also available.